npj: еҸӮж•°еҸҳеҠӣдёәиғҪйҮҸвҖ”еҠ йҖҹжңәеҷЁеӯҰд№ еҠҝеҮҪж•°жһ„е»ә
 ж–Үз« жҸ’еӣҫ
ж–Үз« жҸ’еӣҫ
еҹәдәҺ第дёҖжҖ§еҺҹзҗҶи®Ўз®—ејҖеұ•жңәеҷЁеӯҰд№ жқҘи®ӯз»ғз»ҸйӘҢеҠҝеҮҪж•°жҳҜиҝ‘е№ҙжқҗж–ҷи®Ўз®—йўҶеҹҹжңҖйҮҚиҰҒзҡ„иҝӣеұ•д№ӢдёҖ гҖӮ зІҫзЎ®и®ӯз»ғзҡ„еҠҝеҮҪж•°е…·жңүеҸҜд»ҘжҜ”жӢҹ第дёҖжҖ§еҺҹзҗҶзҡ„зІҫеәҰ пјҢ дҪҶи®Ўз®—йҮҸжҳҫи‘—йҷҚдҪҺ пјҢ е…¶и®Ўз®—йҮҸд»…дёҺжЁЎжӢҹеҺҹеӯҗж•°жҲҗзәҝжҖ§е…ізі» пјҢ иҖҢе…ёеһӢзҡ„еҜҶеәҰжіӣеҮҪи®Ўз®—дёҖиҲ¬дёҺеҺҹеӯҗж•°жҲҗдёүж¬Ўж–№е…ізі» гҖӮ еӣ жӯӨ пјҢ еҹәдәҺжңәеҷЁеӯҰд№ з»ҸйӘҢеҠҝеҸҜд»Ҙз”ЁжқҘй«ҳзІҫеәҰең°жЁЎжӢҹи¶…еӨ§и§„жЁЎзҡ„еӨҚжқӮжқҗж–ҷдҪ“зі» гҖӮ еҮҶзЎ®зҡ„жңәеҷЁеӯҰд№ з»ҸйӘҢеҠҝйңҖиҰҒеҗҢж—¶еҲ©з”Ёз¬¬дёҖеҺҹзҗҶиҺ·еҫ—зҡ„еҠӣе’ҢиғҪйҮҸжқҘеҗҢж—¶и®ӯз»ғ гҖӮ з”ұдәҺеҠӣжҳҜиғҪйҮҸзҡ„дёҖйҳ¶еҜјж•° пјҢ иҝҷеҜјиҮҙйңҖиҰҒеҚҒеҲҶеәһеӨ§зҡ„ж•°жҚ®йӣҶжүҚиғҪе®һзҺ°еҮҶзЎ®зҡ„и®ӯз»ғз»“жһң пјҢ еҗҢж—¶еҜ№и®ӯз»ғж–№жі•д№ҹжҸҗеҮәеҫҲеӨ§жҢ‘жҲҳ гҖӮ
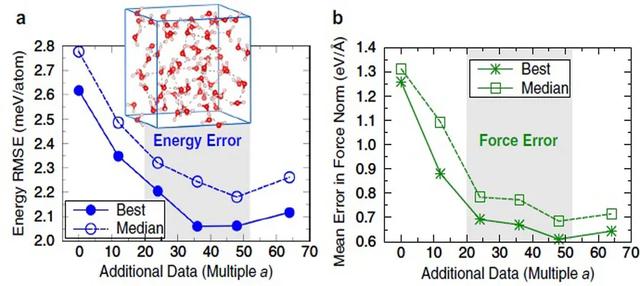
жқҘиҮӘзҫҺеӣҪгҖҒеҫ·еӣҪе’ҢиӢұеӣҪзҡ„иҒ”еҗҲз ”з©¶еӣўйҳҹ пјҢ жҸҗеҮәе°ҶиғҪйҮҸз”ЁеҺҹеӯҗй—ҙдҪңз”ЁеҠӣжі°еӢ’еұ•ејҖ пјҢ еӣ жӯӨе·§еҰҷең°з»•ејҖдәҶдёҠиҝ°й—®йўҳ гҖӮ еҹәдәҺжі°еӢ’еұ•ејҖзҡ„иғҪйҮҸиЎЁиҫҫејҸ пјҢ 他们жҸҗеҮәдәҶж–°зҡ„еҠҝеҮҪж•°и®ӯз»ғж–№жі• гҖӮ и®Ўз®—з»“жһңиЎЁжҳҺ пјҢ зӣёжҜ”зӣҙжҺҘйҮҮз”ЁдҪңз”ЁеҠӣи®ӯз»ғеҠҝеҮҪж•°зҡ„ж–№жі• пјҢ иҜҘж–№жі•дёҚд»…е°ҶеҠҝеҮҪж•°зҡ„зІҫеәҰжҸҗеҚҮдәҶ50% пјҢ еҗҢж—¶и®Ўз®—ж•ҲзҺҮжҳҺжҳҫжҸҗеҚҮ гҖӮ дёәиҝӣдёҖжӯҘйӘҢиҜҒиҜҘж–№жі•зҡ„жңүж•ҲжҖ§ пјҢ дҪңиҖ…йҖүеҸ–дәҶдёүдёӘе…·дҪ“зҡ„жЁЎеһӢдҪ“зі» пјҢ еҚіж°ҙеҲҶеӯҗеӣўз°ҮгҖҒж¶ІжҖҒж°ҙе’ҢеӨҚжқӮйҮ‘еұһж°§еҢ–зү©ејҖеұ•еҠҝеҮҪж•°и®ӯз»ғ гҖӮ 他们еҸ‘зҺ° пјҢ иҜҘж–№жі•еҸҜд»Ҙжҳҫи‘—йҷҚдҪҺи®ӯз»ғжүҖйңҖзҡ„ж•°жҚ®йӣҶеӨ§е°Ҹпјӣе…·жңүеҫҲеҘҪзҡ„еҸҜ移жӨҚжҖ§ пјҢ еҸҜд»ҘеҮҶзЎ®йў„жөӢж•°жҚ®йӣҶд№ӢеӨ–зҡ„ж–°дҪ“зі»пјӣеҗҢж—¶еҸҜд»ҘжҸҗеҚҮдҪңз”ЁеҠӣйў„жөӢзҡ„еҮҶзЎ®жҖ§ гҖӮ з®ҖиҖҢиЁҖд№Ӣ пјҢ иҜҘж–№жі•з®ҖеҢ–дәҶзҘһз»ҸзҪ‘з»ңз»ҸйӘҢеҠҝзҡ„жһ„йҖ иҝҮзЁӢ пјҢ жңүжңӣжҺЁе№ҝеә”з”ЁдәҺд»»ж„Ҹзұ»еһӢзҡ„жқҗж–ҷдёӯ гҖӮ
гҖҗnpj: еҸӮж•°еҸҳеҠӣдёәиғҪйҮҸвҖ”еҠ йҖҹжңәеҷЁеӯҰд№ еҠҝеҮҪж•°жһ„е»әгҖ‘иҜҘж–Үиҝ‘жңҹеҸ‘иЎЁдәҺnpj Computational Materials 6: 54 (2020) пјҢ иӢұж–Үж ҮйўҳдёҺж‘ҳиҰҒеҰӮдёӢ пјҢ зӮ№еҮ»еҸҜд»ҘиҮӘз”ұиҺ·еҸ–и®әж–ҮPDF гҖӮ
 ж–Үз« жҸ’еӣҫ
ж–Үз« жҸ’еӣҫ
Efficient training of ANN potentials by including atomic forces via Taylor expansion and application to water and a transition-metal oxide
April M. Cooper, Johannes K?stner, Alexander Urban & Nongnuch Artrith
Artificial neural network (ANN) potentials enable the efficient large-scale atomistic modeling of complex materials with near first-principles accuracy. For molecular dynamics simulations, accurate energies and interatomic forces are a prerequisite, but training ANN potentials simultaneously on energies and forces from electronic structure calculations is computationally demanding. Here, we introduce an efficient alternative method for the training of ANN potentials on energy and force information, based on an extrapolation of the total energy via a Taylor expansion. By translating the force information to approximate energies, the quadratic scaling with the number of atoms exhibited by conventional force-training methods can be avoided, which enables the training on reference datasets containing complex atomic structures. We demonstrate for different materials systems, clusters of water molecules, bulk liquid water, and a lithium transition-metal oxide that the proposed force-training approach provides substantial improvements over schemes that train on energies only. Including force information for training reduces the size of the reference datasets required for ANN potential construction, increases the transferability of the potential, and generally improves the force prediction accuracy. For a set of water clusters, the Taylor-expansion approach achieves around 50% of the force error improvement compared to the explicit training on all force components, at a much smaller computational cost. The alternative force-training approach thus simplifies the construction of general ANN potentials for the prediction of accurate energies and interatomic forces for diverse types of materials, as demonstrated here for water and a transition-metal oxide.
жҺЁиҚҗйҳ…иҜ»

















- й»‘йІЁ4proд»Җд№Ҳж—¶еҖҷеҮәеӨҡе°‘й’ұпјҢй»‘йІЁ4proд»·ж јеҸӮж•°д»Ӣз»Қ
- зәўзұіk40proе’Ңiqooneo3е“ӘдёӘеҘҪжҖ§д»·жҜ”й«ҳ еҸӮж•°еҜ№жҜ”еҢәеҲ«иҜ„жөӢ
- дёҖеҠ йҰ–ж¬ҫеҸҜз©ҝжҲҙи®ҫеӨҮOnePlus Bandж¶ҲжҒҜжұҮжҖ»пјҡд»·ж јгҖҒеҸӮж•°гҖҒеҠҹиғҪе…ЁжҸӯз§ҳ
- иҚЈиҖҖV40жӯЈејҸеҫ—еҲ°зЎ®и®ӨпјҒеҸӮж•°й…ҚзҪ®д№ҹеҹәжң¬зЎ®е®ҡпјҒе”®д»·жҲ–е°ҶжҳҜжғҠе–ң
- еҚҺдёәз•…дә«20seе’Ңзәўзұіnote9е“ӘдёӘеҘҪеҢәеҲ«еңЁе“Ә еҸӮж•°еҜ№жҜ”иҜ„жөӢ
- realmev15е’Ңrealmev3еҢәеҲ«еҸӮж•°еҜ№жҜ” е“ӘдёӘеҘҪжҖ§д»·жҜ”й«ҳ
- зәўзұіk40proе’ҢиҚЈиҖҖ30еҢәеҲ«е“ӘдёӘеҘҪ дёҚеҗҢзӮ№еҜ№жҜ”еҸӮж•°й…ҚзҪ®и°ҒеҘҪ
- iqoo7е’Ңзәўзұіk30иҮіе°ҠзәӘеҝөзүҲе“ӘдёӘеҘҪеҢәеҲ«еңЁе“Ә еҸӮж•°еҜ№жҜ”иҜ„жөӢ
- е°Ҹзұі11е’Ңзәўзұіk30proе“ӘдёӘеҘҪжҖ§д»·жҜ”й«ҳ еҸӮж•°й…ҚзҪ®еҜ№жҜ”еҢәеҲ«
- еҚҺдёәnova8дёҺе°Ҹзұі10еҜ№жҜ”е“ӘдёӘеҘҪ еҸӮж•°й…ҚзҪ®еҢәеҲ«жҖ§иғҪиҜ„жөӢ